Cardiovascular disease (CVD) including heart disease, vascular disease and atherosclerosis are the most critical global health threats.
An estimated 26 million people are living with the effects of heart disease and is a major cause of death in western society. Until recently the widely held belief was that the CVD is simply the process as a build up of fat on the surface of artery walls. Eventually, this build up of fat blocks the artery and a heart attack or stroke occurs. However, the process has now been identified as a disease of the inner artery wall (intima) and inflammation is a key factor in its progression.
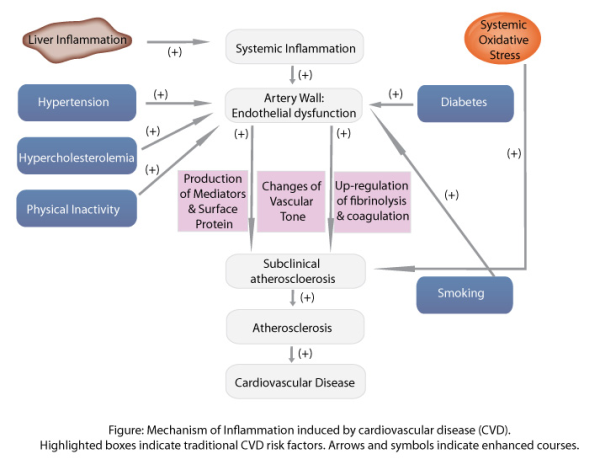
The source of inflammation in CVD is not completely understood. However, numerous factors are thought to initiate the complex inflammatory process such infectious agents for example herpes viruses and Chlamydia pneumoniae. Other promoters and stimulators of inflammation leading to endothelial injury include smoking, hyperglycaemia, oxidised low-density lipoprotein (LDL) or sheer stress on the vessel wall by hypertension. Genetic factors may also play a role in the degree and duration of the inflammatory response, although this still needs to be fully explored.
Once stimulated by a promoter or stimulator (including those mentioned above), endothelial cells of the intima interpret their presence as unwanted and activate the immune system to deal with the problem. The gene transcription factor NF-kB is released, serving as a promoter of early cytokines such as TNF-α and IL-6, chemokines such as MCP-1 and adhesion molecules. The chemokines attract monocytes and T lymphocytes (T cells) from the blood stream allowing monocytes to travel across the endothelial barrier and become macrophages. Entry of monocytes into the vessel wall is a key factor in the development of atherosclerosis, as blocking monocyte migration has ameliorated atherosclerosis in in vivo models (1). Once inside the intima, these mononuclear cells produce pro-inflammatory cytokines such as IL-1, IL-6 and TNF-α to stimulate the inflammatory cascade. Metalloproteinases are also released, promoting smooth muscle cell proliferation and uptake of LDL by these macrophages to form foam cells.
Through uptake of LDLs, a fatty streak can develop into a necrotic plaque that is sealed off from the blood flow by the fibrous cap and is held in balance by collagen deposition and degredation. Fissuring or rupturing of this cap can occur when the balance is disrupted by increased inflammation leading to thinning of the collagen cap. The plaque rupture exposes thrombotic substances to the blood, leading to local thrombus formation and downstream microemobolization (2). Furthermore, inflammatory cytokines activate platelets expressing P-selectin and CD40, thus increasing platelet-platelet adhesiveness (3). Cytokines also signal the production of acute phase proteins such as fibrinogen serum amyloid A and C-reactive protein. These are systemic downstream markers which can be useful in assessing cardiovascular risk in patients.
The role of inflammation in cardiovascular disease is not strictly limited to the innate inflammatory response. The adaptive immune response particularly lymphocytes are also involved in CVD. Flow cytometry based methods have quantitatively investigated the cell composition of a normal aortas (4, 5). These have demonstrated that both T and B lymphocytes, macrophages and dendritic cells reside within a major site of the arterial wall (lamina adventitia) of non inflamed aortas. To further visualise the induction of the immune response and investigate the relationship between the immune and cardiovascular systems, multiphoton laser-scanning microscopy (MPLSM) could be used, however this is still at a method development stage (6).
Prevention of the initial development of CVD and progression over time is the goal of any prevention program. With increasing knowledge, the approach to identifying the underlying causes of heart disease is changing rapidly. Much research has identified inflammation as an underlying or active factor in the development of the disease. For the past two decades, clinical trials of antiatherosclerotic drug therapies have sought to reduce CVD morbidity and mortality. This includes the use of a group of drugs called statins (atorvastatin and rosuvastatin) (7) to treat high cholesterol levels which have been shown in large randomised trials, to reduce cardiovascular events in risk patients (8). Research has demonstrated that at higher doses, statins slow or even reverse plaque progression as demonstrated during intravascular ultrasound (9). Recently however, clinical findings have indicated that statins may slow progression of disease at a rate and to an extent that cannot be attributed to lower LDL alone. The proposed mechanisms for such pleiotropic actions include endothelial-dependent nitric oxide bioavailability, inhibition of oxidative stress and anti-inflammatory activity. In particular a number of clinical trials have shown that statins reproducibly lower circulating levels of C reactive protein (CRP) an inflammatory biomarker associated with acute coronary syndromes (10). Reducing inflammation may therefore be a key mechanism by which statins alter the biology of the plaque and slow down disease progression.
Although statins are currently the most popular and widely prescribed drugs to help treat CVD, evidence indicates side effects such as a higher risk of drug interactions in elderly, muscle pain or memory related problems are linked to their use. It is therefore necessary to continue the investigation into inflammation and in inflammatory cell-cell interactions to help develop more effective therapies.
![]()
References
- Stewart SH, Mainous AG III, Gilbert G. J Am Board Fam Pract 2002;15:437-442.
- Taylor, Marcia L. Southern Medical Journal 2004.
- Mainous AG, Pearson WS. Fam Med 2003;35:112-118.
- Galkina E, Kadl A, Sanders J, Varughese D, Sarembock IJ, Ley K. J Exp Med. 2006; 203: 1273–1282.
- Jongstra-Bilen J, Haidari M, Zhu SN, Chen M, Guha D, Cybulsky MI. J Exp Med. 2006; 203: 2073–2083.
- Owain R. Millington, James M. Brewer, Paul Garside and Pasquale Maffia. Methods In Molecular Biology. 2010; 616: part 3 193-206.
- http://en.wikipedia.org/wiki/Statin.
- Jain MK, Ridker PM: Nat Rev Drug Discov, 2005; 4: 977-987.
- Ridker PM, Danielson E, Fonseca FA, Genest J, Gotto AM, Jr., Kastelein JJ, Koenig W, Libby P, Lorenzatti AJ, MacFadyen JG, Nordestgaard BG, Shepherd J, Willerson JT, Glynn RJ: N Engl J Med, 2008; 359: 2195-2207.
- Nissen SE, Tuzcu EM, Schoenhagen P, Crowe T, Sasiela WJ, Tsai J, Orazem J, Magorien RD, O'Shaughnessy C, Ganz P: N Engl J Med, 2005; 352: 29-38.





