Scleroderma is complex and highly heterogeneous group of connective tissue disorders that are characterized by widespread tissue fibrosis, immunologic abnormalities, alterations to the vasculature and production of autoantibodies against various cellular antigens [1]. The disease manifests itself clinically to various degrees, from limited cutaneous involvement affecting localized regions of the hands, arms and face, to systemic fibrotic disease involving the esophagus, heart, lungs, kidneys, GI tract, and liver (also known as systemic sclerosis). Although its pathogenesis has not been fully elucidated, the disease results in vascular injury to endothelial cells (ECs) and widespread chronic inflammation, ultimately leading to excessive accumulation of extracellular matrix proteins, namely collagens type I, type III, type V and Fibronectin [2-4 ].
While estimates vary, prevalence likely ranges between 50 to 300 cases per 1 million persons, with women at increased risk over men (5). Familial clustering of disease suggests a potential role for genetic risk factors, with scleroderma-associated gene polymorphisms being reported for cytokines, cytokine receptors, chemokines and extracellular proteins [6]. However, environmental factors, such as viral infection or exposure to silica and poly vinyl, can induce phenotypes that are similar to scleroderma and may play a role in the disease [7]. After adjusting for age and gender differences in disease outcomes, mortality rates in systemic scleroderma are estimated to be 5 to 8 times greater than mortality risks of the general population, and mortality risk is highest in patients with the greatest degree of multi-organ involvement [7]. Survival has improved over the past few decades, likely due to increased detection and development of improved anti-inflammatory drugs, with average 5-year survival rates post-diagnosis of between 70-80% [8]. Death often results from complications of fibrotic disease in various organ systems, commonly gastrointestinal events, interstitial lung disease with pulmonary hypertension and cardiovascular disease.
Scleroderma patients have been shown to have significant immunological abnormalities such as increased T-cell activation, altered T-cell ratios and increased levels of dermal macrophage [9]. Both the adaptive and innate immune systems have been implicated in progressive disease, with contributions from pro-inflammatory cytokines and activation of pro-fibrotic pathways in endothelial cells and fibroblasts. Extended recruitment and activation of fibroblasts leads to progressive structural damage of capillaries, resulting in capillary atrophy and vasculopathies such as Renaud’s Phenomenon [10, 11].
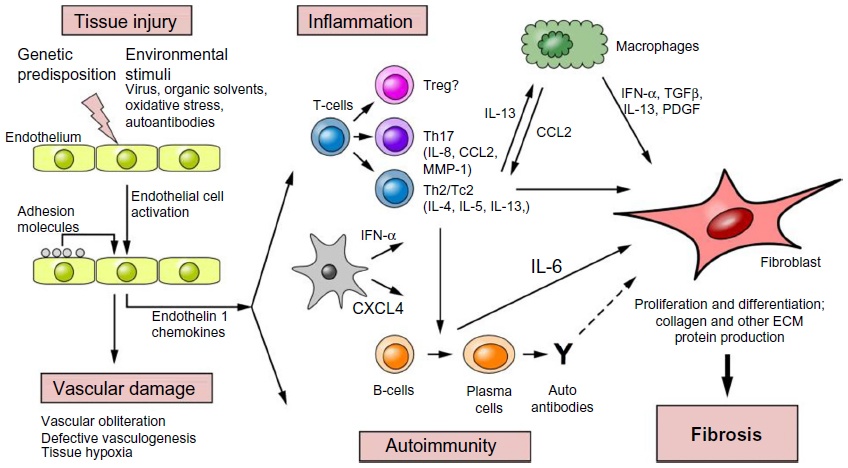
(Above Illustration taken from Fuschiotti 2016 [17], Figure 1)
With no currently approved FDA therapies to directly treat scleroderma, there is a great unmet medical need for development of efficacious therapies. Preclinical animal models have been instrumental in understanding the pathogenesis of the disease, however no model entirely recapitulates the pathophysiology of the human disease course. Several spontaneous genetic models exist that mimic certain aspects of the disease, such as the Tsk1/+ and Tsk2/+ mouse models, however mutations associated with these strains do not appear to be highly relevant to the human disease [12]. The UCD-200 chicken model has been studied broadly and may be the most relevant spontaneous animal model of the human disease, demonstrating late stage perivascular infiltrate, vascular occlusion with endothelial cell injury, nuclear auto-antibodies as well as fibrosis of the skin and visceral organs [12]. Genomic studies of this model have demonstrated association with five genes of interest, including TGFBR1, IGFBP3, EOXC2/IRF4, CCR8 and SOCS1, all of which have been implicated in autoimmune conditions. [13].
Of the inducible fibrotic disease models available, by far the best characterized is the bleomycin-induced murine model of fibrosis. Bleomycin is a chemotherapeutic antibiotic isolated from the bacterium Steptomyces verticillius, and is a known inducer of fibrosis in multiple species including mice, rats, dogs and humans [14]. This model encompasses many features of human disease, including inflammation and autoimmune attack, vasculopathy and fibrosis of the skin and organs. Following administration, an inflammatory cascade is quickly initialized consisting of pro-inflammatory cytokines such as IL-1, TNF-alpha, IL-6, and IFN-gamma and is followed closely thereafter by the pro-fibrotic markers TGF-beta, fibronectin, and procollagen-1 [15]. Strain susceptibility varies greatly however, with high susceptibility in CBA/J and C57BL/6 mice whereas Balb/c are fibrosis-resistant [16]. Multiple bleomycin administration methods have been utilized, including intra-tracheal administration for development of pulmonary fibrosis, repeated localized injections for dermal fibrosis, and, more recently, multisite injections over an extended period of time to develop disease that is more systemic in nature. These administration methods however are difficult to standardize and replicate reproducibly, and so newer techniques involve continuous administration of bleomycin utilizing implanted osmotic minipumps for an extended and controlled delivery [8].
MD Biosciences has validated this osmotic minipump delivery method of bleomycin to model scleroderma. Results of this validation, including multiplex cytokine profiling, multi-site histologic analysis and additional fibrotic assessments, are now available. This model is being used for therapeutic programs with indications in both scleroderma as well as fibrotic disease more broadly.
For sponsors interested in fibrotic disease studies with MD Biosciences, please contact us to request additional information or to speak with a scientist.

References:
1) Gabrielli, A., Avvedimento, E. V. & Krieg, T. Scleroderma. N. Engl. J. Med. 360, 1989–2003 (2009).
2) Perlish J.S., Lemlich G., and Fleischmajer R. 1988. Identification of collagen fibrils in scleroderma skin. J. Invest. Dermatol. 90:48–54. 10.1111/1523-1747.ep12462561
3) Martin P, Teodoro WR, Velosa AP, de Morais J, Carrasco S, Christmann RB, et al. Abnormal collagen V deposition in dermis correlates with skin thickening and disease activity in systemic
sclerosis. Autoimmun Rev (2012) 11:827–35.10.1016/j.autrev.2012.02.01
4) Mauch C, Eckes B, Hunzelmann N, Oono T, Kozlowska E, Krieg T (1993) Control of fibrosis in systemic scleroderma. J Invest Dermatol 100:92–96
5) Chifflot H, Fautzi B, Sordet C, Chatelus E, Sibilia J. Incidence and prevalence of systemic sclerosis: a systematic literature review. Semin Arthritis Rheum 2008; 37:223-35.
6) Agarwal SK, Tan FK, Arnett FC. Genetics and genomic studies in scleroderma (systemic sclerosis). Rheum Dis Clin North Am 2008;34:17-40.
7) Nietert PJ, Silver RM. Systemic sclerosis: environmental and occupational risk factors. Curr Opin Rheumatol 2000;12: 520-6
8) Soumya Chatterjee. Systemic scleroderma. Cleveland clinic -center for continuing education. Published: August 2010.
9) Liang M, Lv J, Zou L, Yang W, Xiong Y, Chen X, et al. A modified murine model of systemic sclerosis: bleomycin given by pump infusion induced skin and pulmonary inflammation and fibrosis. Lab Invest. 2015;95(3):342–50.
10) Gilbane A. J., Denton C. P., Holmes A. M. Scleroderma pathogenesis: A pivotal role for fibroblasts as effector cells. Arthritis Research & Therapy. 2013;15(3, article no. 215)
11) Y. Mostmans, M. Cutolo, C. Giddelo et al., “The role of endothelial cells in the vasculopathy of systemic sclerosis: a systematic review,” Autoimmunity Reviews, vol. 16, no. 8, pp. 774–786, 2017.
12) Artlett CM. Animal models of systemic sclerosis: their utility and limitations. Open Access Rheumatol. 2014;6: 65–81.
13) Radstake TR, Gorlova O, Rueda B, et al. Genome-wide association study of systemic sclerosis identifies CD247 as a new susceptibility locus. Nat Genet. 2010;42:426–429.
14) Blum RH, Carter SK, Agre K. A clinical review of bleomycin—a new antineoplastic agent. Cancer 1973; 31:903–914.
15) Moeller A., Ask K., Warburton D., Gauldie J., Kolb M. (2008). The bleomycin animal model: a useful tool to investigate treatment options for idiopathic pulmonary fibrosis? Int. J. Biochem. Cell Biol. 40, 362–382.
16) Phan SH, Kunkel SL. Lung cytokine production in bleomycin-induced pulmonary fibrosis. Exp Lung Res. 1992;18(1):29–43.
17) Fuschiotti P. Current perspectives on the immunopathogenesis of systemic sclerosis. Immunotargets Ther. 2016;5:21–35.





