Continuing the discussion of imaging technologies, this week we will cover biofluorescence and bioluminescence as readouts for RA models.
Continuing the discussion of imaging technologies, this week we will cover biofluorescence and bioluminescence as readouts for RA models.
Topics: Inflammation
Rheumatoid arthritis is a chronic and progressive inflammatory condition estimated to affect between 0.5% and 1% of the world’s population, with more women being affected than men. RA is a systemic disease manifesting mainly as a disabling destruction of the synovial joints of the hands and feet. In addition to the disability and decreased quality of life caused by RA, patients are at increased risk of developing cardiovascular disease. Joint destruction is induced by dysregulated immune activation of both the innate and adaptive immune responses resulting in alterations in the synovium, cartilage and bone. The normal joint has a thin synovial lining (intimal lining layer), 1-3 cells thick. Beneath this is a sub-lining layer of connective tissue scattered with immune cells, blood vessels and nerve cells. Together these layers form the synovium, which produces the synovial fluid that serves to lubricate the joint. Disease initiation results in profound changes in the structure and composition of the synovium and synovial fluid; with the infiltration of inflammatory cells, synovial cell hyperplasia, increased angiogenesis, fibroblast proliferation and extracellular matrix production. This increase in synovial cell proliferation can result in the lining increasing up to five times its original size and can result in pannus formation. The culmination of these events is bone and cartilage erosion and loss of joint function.
Topics: Inflammation
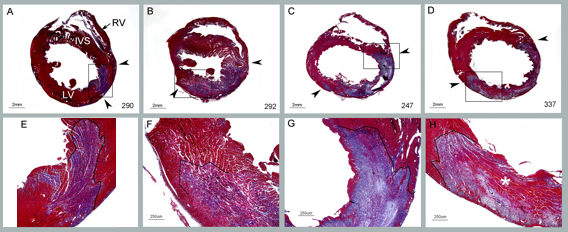
The preclinical occlusion-induced myocardial infarct model is a well-known technique for investigating the cardio-protection of a drug therapy in the event of ischemia/reperfusion injury. The advantage of the model is the ability to study the functional relevance of a drug treatment on the heart following direct coronary flow and the mechanisms by which the drug promotes myocardial protection.
Topics: Cardiovascular
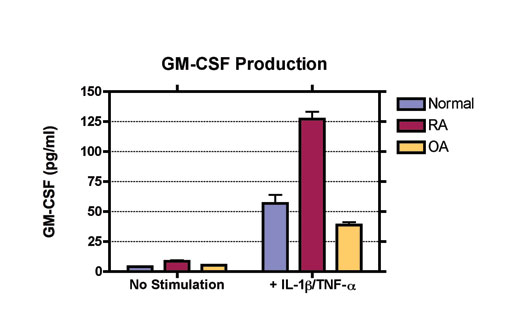
Rheumatoid arthritis (RA) is a chronic autoimmune joint disease characterized by inflammation of the synovium and destruction of cartilage and bone. During synovial inflammation, inflammatory cells (macrophages, mast cells, dentritic cells and lymphocytes) are recruited while resident cells (fibroblast synoviocytes, chondrocytes, osteoclasts, and osteoblasts) are altered to support the inflammatory process. Together, these events create a pathological tissue response.
The synovium consists of two layers, the sublining and intimal lining. In RA, the sublining becomes infiltrated with mononuclear cells, B lymphocytes produce autoantibodies, blood vessels proliferate, lymphoid aggregates form and the intimal lining shows increased cellularity. Macrophages in the synovium produce pro-inflammatory cytokines, chemokines and growth factors which in turn activate fibroblast-like synoviocytes (FLS) to produce their own array of mediators (e.g. proteolytic enzymes, chemokines and cytokines). This produces a paracrine/autocrine network that leads to synovitis, the recruitment of new cells and the destruction of the extracellular matrix. Fibroblast-like synoviocytes have emerged as key pro-inflammatory cells promoting the disease, largely due to their ability to produce massive amounts of degradative enzymes.
The availability of biological therapies has improved clinical outcomes by decreasing inflammation and joint destruction, however only about half of the patients exhibit substantial efficacy. Targeting FLS may further improve clinical outcomes without suppressing systemic immunity. In vitro FLS assays can be used to evaluate effective therapies for arthritis. Using FLS obtained from normal, RA and OA patients, we can evaluate a compound's effect on the production of pro-inflammatory mediators in a preclinical in vitro model.
Topics: Inflammation

We read an interesting article published this week in Journal of Immunology (v184 Bottaro & co.) on the efficacy of anti-CD20 therapy in RA. The article highlights the continuing uncertaintity over the mode of action of B-cell directed therapy in Rheumatoid Arthritis (RA) [review of the differing theories is presented in Clin Exp Immunol. 2009 Aug;157(2):191-7].
Topics: Inflammation
Determine immunomodulatory mechanism of action for compounds in indication discovery or respositioning of approved therapies.
Topics: Inflammation





