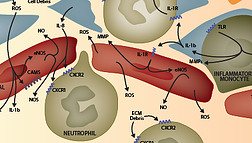
 At the tissue level, inflammation ensues very rapidly after myocardial infarct (MI), initially prompted by detection of high levels of reactive oxygen species (ROS) and necrotic cellular debris by resident cells in neighboring non-infarct tissues. ROS and necrotic cell debris are also detected by peripheral leukocytes, which home to the injured tissue, exit circulation, and infiltrate infarct and non-infarct tissues. Upon entering the lesion site, these leukocytes further release ROS, proteolytic enzymes, pro-inflammatory and cytotoxic diffusible factors and participate in phagocytosis of necrotic cells and disrupted ECM. This post-MI inflammatory environment in cardiac tissues peaks at 1 to 2 weeks and generally resolves at 3 to 4 weeks after the ischemic event. While important for clearing the tissue of compromised cells and debris and preparing it for transitioning into the proliferative phase of infarct healing, inflammation that becomes excessive or chronic results in adverse remodeling, infarct expansion, and poor patient outcomes. [1-4]
At the tissue level, inflammation ensues very rapidly after myocardial infarct (MI), initially prompted by detection of high levels of reactive oxygen species (ROS) and necrotic cellular debris by resident cells in neighboring non-infarct tissues. ROS and necrotic cell debris are also detected by peripheral leukocytes, which home to the injured tissue, exit circulation, and infiltrate infarct and non-infarct tissues. Upon entering the lesion site, these leukocytes further release ROS, proteolytic enzymes, pro-inflammatory and cytotoxic diffusible factors and participate in phagocytosis of necrotic cells and disrupted ECM. This post-MI inflammatory environment in cardiac tissues peaks at 1 to 2 weeks and generally resolves at 3 to 4 weeks after the ischemic event. While important for clearing the tissue of compromised cells and debris and preparing it for transitioning into the proliferative phase of infarct healing, inflammation that becomes excessive or chronic results in adverse remodeling, infarct expansion, and poor patient outcomes. [1-4]
At the cellular level, resident cells including cardiomyocytes, endothelial cells, vascular smooth muscle cells, and fibroblasts that survive initial ischemia-induced necrosis are threatened by extracellular “danger” signals that may induce apoptosis. In response to the abundance of ROS and necrotic cell debris, they draw neutropils and later monocytes/macrophages to the injured area by releasing chemoattractants. Inflammation at the cellular level after MI is characterized by both harmful and helpful processes – continued cell death, release of inflammation-amplifying diffusible bioactive mediators and ROS, and clearing of dead cells, cellular debris, and disrupted ECM and finally release of pro-resolution “stop signals” and anti-inflammatory mediators. [2,4]
Since myocardium generally has only very limited regenerative capability, the space vacated by the death of cardiomyocytes is filled by a fibrotic scar. Therapeutic strategies targeting the cells affected by MI (i.e., resident cells) and the cells responding to MI (i.e., invading cells) offer unique opportunities to influence patient outcomes. New research into the promotion of myocardial regeneration using cell-based therapy strategies, such as stem cell transplantation or stem cell mobilization, appears promising. However, so far most of the positive effects of stem cells appear to result from paracrine actions of the cells on existing tissues rather than differentiation and incorporation of these stem cells at the lesion site. Paracrine signaling of engrafted cells may act by reducing apoptosis, inflammation, and fibrosis, and encouraging angiogenesis and other repair processes. [1,2] At the cellular level, several pharmacological agents are being evaluated for their therapeutic benefits after acute MI. These are generally acting either to directly protect resident cells or dampen the actions of infiltrating cells. Adrenomedullin has demonstrated cardoprotective effects in experimental animal models of I/R injury including suppression of cardiomyocyte apoptosis via interference with the PI3K-Akt cascade and inhibition of ROS production, resulting in reduced infarct size. In clinical studies, Adrenomedullin appears to reduce oxidative stress and reduce infarct size in patients undergoing reperfusion therapy. [5] Compounds that act on signaling molecules in the RISK (signaling pathway compromising the reperfusion-injury salvage kinase) pathway, such as Atrial Natriuretic Peptide (ANP), appear to have cardioprotective effects that include suppression of apoptosis and induction of pro-survival signaling pathways. In ex vivo animal hearts, ANP delivered during reperfusion decreases infarct size. In clinical studies, ANP administered to patients receiving reperfusion therapy appears to guard against ischemic reperfusion (I/R) injury and improve LV function. [5,6] Systemic immunosuppression achieved via drugs such as Methotrexate delivered in combination with reperfusion therapy have been shown to reduce infarct size in animal models. [1] Adenosine, when administered along with reperfusion therapy within 6 hours of MI, reduces infarct size and improves outcomes in experimental animal models and patients. Adenosine is known to inhibit neutrophils and platelets as well as dilate cardiac microvasculature and serve as a substrate for ATP replenishment. [5,6]
At the molecular level, an abundance of bioactive compounds act via autocrine, paracrine, and endocrine signaling to initiate signal transduction cascades that ultimately influence gene transcription. All of these interactions are regulated in space (i.e., different cell types) and time (i.e., after onset of infarct symptoms) to create an initially detrimental and eventually beneficial microenvironment after MI. Although these enormous complexities result in relatively slow progress toward effective therapies, there are innumerable opportunities for targeted therapeutic intervention.
MD Biosciences offers preclinical models for myocardial or I/R drug research. If you'd like to explore a study design, contact us and we'd be happy to discuss a protocol with you.
![]()
- Steffens, S., Montecucco, F., and Mach, F. (2009). Thrombosis and Haemostasis, 102(2), 240-247.
- Jiang, B. and Liao, R. (2010). Journal of Cardiovascular Translational Research, 3(4), 410-416.
- Sun, Y. (2009). Cardiovascular Research, 81(3), 482-490.
- Dobaczewski, M., Gonzalez-Quesada, C., and Frangogiannis, N.G. (2010). Journal of Molecular and Cellular Cardiology, 48(3), 504-511.
- Yasuda, S. and Shimokawa, H. (2009). Circulation Journal, 73(11), 2000-2008.
- Liem, D.A., Honda, H.M., Zhang, J., Woo, D., and Ping, P. (2007). Journal of Applied Physiology, 103(6), 2129-2136.





