Many of the current therapies designed for RA focus on reducing the inflammation present within the joints but do not impact on the process of bone erosion; therefore one of the current goals in RA research is to inhibit the bone destruction that occurs (7). This exciting new field of research is known as osteoimmunology and it is beginning to highlight the link between the immune system and the skeletal system in the development, progression and establishment of RA (8).
The skeletal system consists of bone, cartilage and the connective tissues that connect the bones. Bone comprises of a solid matrix containing hydroxyapatite crystals, collagen fibres and cells. The main types of cells present within the bone are osteocytes, osteoblasts, osteoprogenitor cells and osteoclasts. Osteocytes are mature cells that maintain the bone matrix by dissolving and rebuilding it; osteoprogenitor cells are mesenchymal stem cells that differentiate into osteoblasts; osteoblasts are immature cells that produce new bone matrix, a process known as osteogenesis; and osteoclasts are multinucleated cells of the monocyte/macrophage lineage that degrade bone using hydrochloric acid and enzymes such as cathepsin K and matrix metalloproteinases in a process known as bone resorption (9). In a healthy individual there is a delicate balance between the number and function of osteoblasts and osteoclasts present within the joint, ensuring that in the normal process of bone remodeling the bone that is degraded is replaced. In an individual affected by RA several factors result in an increase in the number and function of osteoclasts, offsetting this balance and causing destructive bone erosion (10).
Interestingly, this field of osteoimmunology is beginning to pinpoint the inflammatory processes present within the arthritic joint, which are driving the osteoclast differentiation and activation. RA was previously thought to be a Th1 mediated disease; however, research has shown that it is most likely that Th17 cells are involved in the pathogenesis of RA, and it has now been established that there is a link between the Th17 cells which are found in the joint and osteoclast function (10,11).
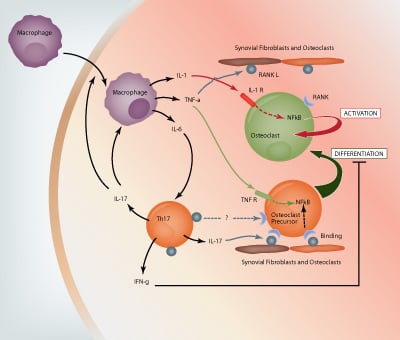
IL-6 along with TGF-β in the presence of IL-23 induces Th17 cell differentiation. IL-6, IL-23 and TGF-β are all produced by macrophages; IL-23 is also produced by activated dendritic cells and TGF-β by synovial fibroblasts (1,3). Th17 cells produce several cytokines including IL-17A, IL-17F, IL-21 and IL-22 (8). IL-17A, which has been found in high concentrations in the synovium and synovial fluid of patients with RA, has multiple functions. It indirectly induces RANKL (receptor activator of NF- κB ligand) expression by synovial macrophages to produce IL-1 and TNF-α, and directly induces expression of RANKL on synovial fibroblasts and osteoblasts (12). RANKL binds to RANK on osteoclast precursor cells and allows these cells to differentiate into mature osteoclasts. Th17 cells also express RANKL, however the current literature shows that Th17 cells alone cannot induce osteoclastogenesis, osteoblasts are also required (11). This may be due to the fact that Th17 cells also produce a low amount of IFNγ, which is known to inhibit the differentiation of osteoclast precursor cells into mature osteoclasts (11).
Along with T cells, macrophages, neutrophils, mast cells and B cells are known t infiltrate the joint and contribute to the ongoing inflammation (1,4). Synovial macrophages express IL-1, IL-6 and TNF-α which are involved in the process of bone resorption (see Figure 1). IL-1 binds to the IL-1 receptors present on mature osteoclasts and TNF-α binds to TNF receptors present on osteoclast precursor cells. Both cytokine-receptor interactions trigger the expression of the transcription factor NF-κB, which allows the activation of osteoclasts and differentiation of precursor cells, respectively (12). TNF-α also induces the expression of RANKL on synovial fibroblasts and osteoblasts, and TNF receptors on osteoclast precursor cells, both of which are important in the differentiation and activation of osteoclasts (8,11,12).
Several interesting developments have already been made within this exciting new field of research. Lubberts et al showed that use of anti-IL-17A in the collagen induced murine model of RA decreased RANKL expression on synovial fibroblasts and osteoblasts and also decreased clinical arthritis scores observed (13); and Sato et al have shown that there is a positive correlation between IL-23 and RANKL expression in the synovium of patients with RA(11). Both studies therefore positively maintain the theory that Th17 cells represent a target for further therapeutic studies in RA. As this new cross-over field highlights further links between the bone remodeling process and the immune system, the prospect for new therapies which aim to tackle the inflammation and the bone erosion in RA looks promising.
References
- McInnes, I.B. and Schett, G. Nature Reviews Immunology. 7, 429-442 (2007).
- Birnbaum, H. et al. Current Medical Research and Opinion. 26(1), 77-90 (2010).
- Brennan, F.M. and McInnes, I.B. The Journal of Clinical Investigation. 118(11), 3537-3545 (2008).
- Cascão, R. et al. Neutrophils in rheumatoid arthritis: Autoimmunity Reviews. 9, 531-535 (2010).
- Brand, D.D. et al. Springer Seminars in Immunopathology. 25, 3-18 (2003).
- Nandakumar, K.S. and Holmdahl, R. Arthritis Research and Therapy. 8, 223 (2006).
- van Vollenhoven, R.F. Nature Reviews Rheumatology. 5, 531-541 (2009).
- Okamoto, K. and Takayanagi, H. International Immunopharmacology (2010). doi: 10.1016/j.intimp.2010.11.010
- Martini, F.H. Fundamentals of Anatomy and Physiology, 7th Edition.
- Sato, K. Allergology International. 57, 109-114 (2008).
- Sato, K. et al. The Journal of Experimental Medicine. 203 (12), 2673-2682 (2006).
- Adamopoulos, I.E. and Bowman, E.P. Arthritis Research and Therapy. 10, 225 (2008).
- Lubberts, E. et al. Arthritis and Rheumatism. 50(2), 650-659 (2004).





